At a Glance
- Gene therapy for inherited eye diseases of the eye is conceptually attractive, but has drawbacks relating to vector characteristics and health economics
- Now, innovative vectors both circumvent the transgene silencing issue associated with legacy non-viral vectors and avoid the capacity limitations, immunogenicity and insertional mutagenesis potential of viral vectors
- At the same time, new classes of therapeutics – including nonsense suppressors – promise the ability to ameliorate multiple inherited diseases with a single drug
- Together, these advances are likely to provide patients with new and effective options and to transform both the management of genetic disease and the economics of the gene therapy sector.
Anyone who doubts that we have entered the era of genomic medicine should consider the 100,000 Genome Project launched by the Department of Health in 2012. The task of this initiative was to sequence the entire genome of thousands of NHS cancer and rare disease patients; the aim was that whole genome sequencing will give these patients the genetic diagnoses that current tests cannot. Accurate diagnoses in turn will guide treatment and support the development of new therapies – not least, gene therapies. These developments give hope to patients with inherited disease.
But is such hope well-founded? Certainly, gene therapy for genetic eye conditions has advanced in recent years; however, it remains imperfect. Consider the vector – that element of the gene therapy which encapsulates the therapeutic DNA, transports it into the target cell and enables its expression. Most vectors are based on modified viruses; but these have a number of problems. First, their carrying capacity is rather limited – about 5 kilobases (kb) in the case of adeno-associated viral (AAV) vectors used in most current trials. And for those seeking treatments for genetic diseases of the eye, this is a problem, because many inherited retinal diseases are caused by mutations in much larger genes. For example, Type 2 Usher Syndrome – the commonest cause of deaf blindness worldwide – is associated with mutations in USH2A, whose transcript is about 19 kb in length. Viral vectors can’t handle genes of that size, so Usher syndrome patients have no prospect of a gene therapy based on conventional vectors. Similarly, ABCA4 (just over 7 kb) and EYS (10 kb)– the genes behind Stargardt’s Disease and one of the autosomal recessive forms of retinitis pigmentosa, respectively – are too large for standard vectors.
But if the viral vector can accommodate the therapeutic sequence, would all our vector problems be solved? Well, no. Remember, these vectors are based on particles which our immune system is adept at rejecting; any viral proteins associated with the vector may cause inflammation and specific immune responses. And that has consequences; inflammation requires modulation with steroids before, during and after surgery, while the risk of unpredictable consequences from an anamnestic immune response to the vector may preclude repeat administrations. If the first dose of a viral vector-mediated gene therapy is insufficiently effective, bad luck; a second dose is currently not allowed. Luxturna is an example of a gene therapy that is limited to a single administration for this reason. And the problems associated with this vector class don’t end there; some viral vectors, such as lentiviruses, insert DNA into the host genome, which carries the risk of insertional mutagenesis, i.e. the disruption of host genes by integration events.
But if we overcame all of these technical considerations, would gene therapy take off? Not necessarily. The field has a fundamental economic problem which must be resolved if the modality is ever to be broadly applied. In brief, the huge cost of developing a gene therapy – tens of millions of pounds – is a commercial investment that must be recouped. As many inherited diseases are very rare, the development costs are spread over smaller numbers of patients – which translates into very high therapy prices. For example, Luxturna costs $850,000 per administration. Multiply this figure by 250 or more – the number of genes that are known to cause inherited retinal disease – and you start to wonder about the financial impact of gene therapies on over-burdened health care systems.
But regardless of these problems, there are patients out there with inherited diseases of the eye, hoping and asking for effective treatments. What can we say to them?
Answer #1: SMARt vectors
I consider that at least part of the future of gene therapy lies with non-viral vectors. These vectors traditionally were thought to be less effective than viral vectors because they tend to be silenced by cellular machinery – they can get genes into the cell, and the genes would be expressed at first, but after a couple of weeks they would be switched off. That problem, however, has been fixed by the discovery of scaffold or matrix attachment regions (S/MARs). These are naturally-occurring DNA sequences that support the structural configuration of chromatin. By incorporating S/MARs downstream of the DNA cargo of non-viral vectors we can get the therapeutic transgene to sit alongside the host DNA – and one effect of this is that the therapeutic DNA is not silenced by the host cell. Furthermore, S/MAR-containing vector sequences are heritable in that host cell division (mitosis) results in replication of both transgene and host DNA; thus, daughter cells also express the transgene. Importantly, this permanent fix is achieved without integration into the host genome; therefore the risk of insertional mutagenesis is greatly reduced as compared with many viral vectors. Other advantages of non-viral vectors include an unlimited cloning capacity – these systems can easily accommodate large genes, and therefore could make gene therapy accessible to a cohort of patients for whom viral vectors can never provide a treatment. Also, as these vectors don’t have viral components, they have less risk of provoking an immune response after injection. In short, they are intrinsically safer.
For all these reasons, I believe that our non-viral, S/MAR-containing vectors should be safer, more effective and more broadly applicable than historical gene therapy vectors. We are actively developing this system with our collaborator Richard Harbottle from the German Cancer Research Centre, Heidelberg; our first target is Type 2 Usher syndrome, which is unsuitable for gene therapy with standard vectors. There’s a lot of work to do, however, as we are still at the stage of in vitro and animal model studies. Nevertheless, given that our vectors provide expression levels equivalent to those associated with viral vectors – up to a year in mouse retina – it seems likely that our technology will soon reach the clinic.
Answer #2: Stop that nonsense
But even non-viral vectors can’t address the intractable problem associated with gene therapy: the huge cost of treatment, which is largely a consequence of the need to develop a unique therapy for each of many very small patient populations. Is there an answer to this problem? I believe so: development of agnostic treatments able to address elements of molecular pathology that are common to many different inherited diseases. We are taking an approach based on the phenomenon of nonsense suppression. This concept was first discovered when investigating the bactericidal effect of aminoglycoside antibiotics, which weaken the specificity of the bacterial ribosomes so that they read the messenger (m) RNA incorrectly and produce jumbled non-functional proteins, the accumulation of which kills the prokaryotes. Aminoglycoside antibiotics and related compounds have a similar but much weaker effect on human eukaryotic ribosomes – and this can be turned to therapeutic advantage.
Briefly, much genetic disease is associated with single nucleotide mutations that have the effect of introducing a premature stop codon into mRNA, thus truncating translation. These are known as nonsense mutations. When a ribosome finds a nonsense mutation in mRNA, it pauses to sample the correct code and then inserts a release factor, thus terminating translation. Under normal circumstances, in less than 1 percent cases the correct amino acid can be inserted instead of a release factor, leading to correction and continued production of full-length protein. The effect of nonsense suppression compounds, such as the aminoglycosides, however, is to modulate the ribosome such that the correction rate is greatly increased. Indeed, in the presence of some compounds, around 25 percent of protein expressed from a mutant gene is normal. Thus, by remarkable good fortune, nonsense suppressors weaken ribosomal specificity in eukaryotes just enough to override the nonsense mutations during a ribosomal pause, but not enough to interfere with the normal protein sequence during regular transcription or effect natural termination codons (which have a readthrough frequency of <0.1 percent). Perfect for addressing genetic disease!
Clearly then, nonsense suppressor compounds could form the basis of drugs that ameliorate the effect of any genetic disease caused by the presence of premature stop codons. The potential is enormous; up to 70 percent of human genetic disease and about a third of inherited retinal disease is associated with nonsense mutations. Indeed, in some diseases – for example, aniridia – 40 percent of patients exhibit premature stop codons in the causative gene. Nonsense suppression therefore may enable us to address a broad range of genetic disease with a single therapy.
Unfortunately, it is not as simple as just giving aminoglycoside antibiotics to patients with inherited disease; in particular, aminoglycoside antibiotics are associated with hearing loss and kidney damage when used over protracted periods, so must be modified to eliminate these side effects. In fact, a medicinal chemist Timor Baasov from Technion, Israel, has reported removal of toxicity-associated domains of the antibiotic while retaining domains associated with nonsense suppression. These designer aminoglycosides are now coming through to clinical trials. Another problem with the nonsense suppression approach is that our cells have a natural surveillance mechanism – nonsense-mediated decay (NMD)– that corrects nonsense mutations at the mRNA level to prevent the build-up of harmful mutant proteins. This raises a problem for nonsense suppressor drugs – they can’t facilitate the production of corrected protein from mutant mRNA if the mutant mRNA is being destroyed by NMD.
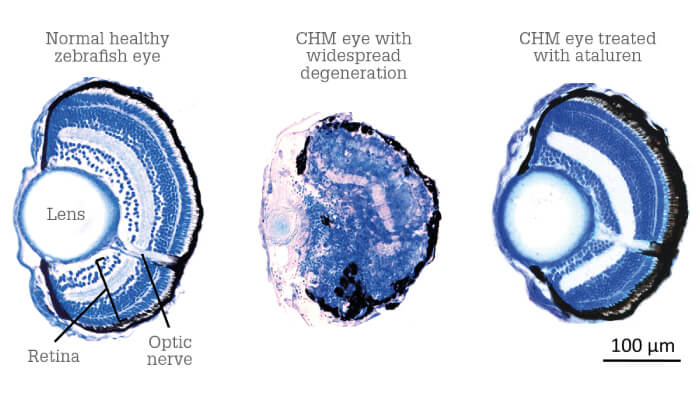
Our nonsense suppression approach, however, addresses both of the above problems. Firstly, we are working with a nonsense suppressor drug candidate that has been used to treat mouth ulcers and asthma for the last 50 years. The non-toxic nature of the compound is therefore well-established, and by ‘repurposing’ an approved drug for indications in genetic disease, we will benefit from a much cheaper and faster development route – the compound has already gone through safety trials, and only requires proof of concept. We’ve already conducted extensive pre-clinical work with this compound, looking at its application for inherited retinal diseases in particular, with excellent results (1). Furthermore, we recently found a possible fix for the issue of nonsense-mediated decay. We’ve shown that every patient has a different level of mutant mRNA – in fact, patients with exactly the same mutation have a 40 percent variation in mRNA levels (2). Checking a patient’s baseline mRNA levels will therefore guide treatment: we can use mRNA levels to predict nonsense suppressor outcomes, and apply drugs, such as caffeine, to boost mRNA levels prior to nonsense suppressor treatment. We are very encouraged by these results and currently investigating the potential of nonsense suppression in conditions including microphthalmia, aniridia, inherited retinal disorders and other genetic eye diseases. Our aim is to design clinical trials that assess our drug not just in one condition, but in perhaps three different inherited retinal diseases associated with nonsense mutations – a more cost-effective approach than running a separate trial for each disease.
In brief, the great promise of nonsense suppression is that it works in a disease- and gene-independent manner, and hence is likely to be highly cost-effective. Furthermore, it could suggest new treatment strategies: for example, children who have started to develop night blindness, but who do not want to immediately opt for invasive gene therapy surgery, could choose nonsense suppression to non-surgically maintain normal protein production and slow retinal degeneration until such time as they are ready for the gene therapy option. It will also potentially help patients with syndromic disease as these drugs can be given orally and therefore improve any systemic dysfunction.
Overcoming economic hurdles
The ideal situation is that every patient with a genetic eye disease receives a genetic diagnosis. But that’s certainly not happening at present; our own research (3) suggests that a patient with genetic eye disease has a 25 percent chance of getting a genetic diagnosis under the current genetic testing regime. The rate rises to about 60 percent for the inherited retinal diseases subset – but what about the other 40 percent? My hope is that increased availability of whole genome sequencing will significantly improve rates of genetic diagnosis for all patients – and turn many of them into candidates for genetic therapy. With regard to implementation of gene therapy approaches, the biggest obstacle remains the economic challenge. The cost of gene therapy development is huge, and given that in some diseases there may be only 100 patients worldwide, it’s hard to see how those costs would be recouped under current circumstances.
Nevertheless, circumstances are changing, and I am confident that the next two to five years will see many more gene therapies entering later stage clinical trials. Most of these will be based on viral vectors. Within five years, however, I expect to also see clinical studies of therapies based on non-viral vectors. Some of these, I hope, will be approved, which will demonstrate the utility of this modality and offer hope to all patients with inherited disorders – not just those whose diseases are compatible with the limitations of viral vectors. Importantly, an increase in the number of approved treatments – not just more gene therapies, but also nonsense suppressors and other therapies – will drive down treatment costs through competition. The advent of therapeutic systems that target common disease pathways, such as those involved in metabolism or cell death, could at least slow down disease progression in patients with inherited retinal disease and prevent further deterioration. One possibility is suggested by Jose Alain-Sahel’s work on rod-derived cone viability factor (RdCVF). This neuroprotectant facilitates glucose uptake in cones, thereby helping them maintain the high metabolic rate necessary for their function. Treating the retina with RdCVF therefore may preserve cones and maintain the central vision of patients at risk of cone loss.
In conclusion, I expect that the combination of existing gene therapy approaches, new vectors and novel modalities, such as nonsense suppressors and metabolic support agents, will provide the basis of an armory that will give patients a new and broad range of effective options. Isn’t that far better than expecting them to rely on one option that does not work in all patients, or which must be discontinued after a small number of administrations, or which is unacceptable to the patient? Being able to offer a significant number of effective alternatives to patients with inherited retinal disease – that’s my dream.
