
- AMD is typically viewed as a single, complex disease entity
- Recent clinical, genetic, histological and functional studies challenge this concept, and suggest that AMD may comprise at least two or three distinct biological diseases
- This implies that a patient may have more than one disease and that treatments should be tailored to specific diseases
- A large, well-characterized cohort of AMD cases and controls, an extensive collection of donated eyes and robust population-based databases are critical resources for assessing this concept
- These resources are being employed to delineate the biology of disease in specific clinical and genetic patient populations
- The resulting knowledge will help to identify disease-specific pathways and targets for drug development efforts
Age-related macular degeneration (AMD) is a leading cause of blindness worldwide.Recently, converging scientific and clinical data have enabled remarkable advances in our understanding of its genetic and biological foundations (see Figure 1 for the pathophysiology of AMD). Aberrant function of the complement system at the level of the retinal pigment epithelium (RPE)-choroid interface plays a key role in AMD disease etiology. This was discovered in early morphological and immunohistochemical studies of human donor eyes. Subsequent genetic analyses identified significant associations with a number of complement pathway-associated genes: a specific common haplotype of the complement factor H (CFH) gene on chromosome 1, which accounts for a significant proportion of risk for developing AMD (1-5); a pair of common protective haplotypes (6-8), one of which – a large deletion distal to CFH – encompasses the CFH-related 1 (CFHR1) and CFHR3 genes; and variations in other complement regulators, including complement factor B (CFB) (9) and complement component 3 (C3) (10). Taken together, these complement-related variations account for a major proportion of the genetic risk for AMD in the Caucasian population.
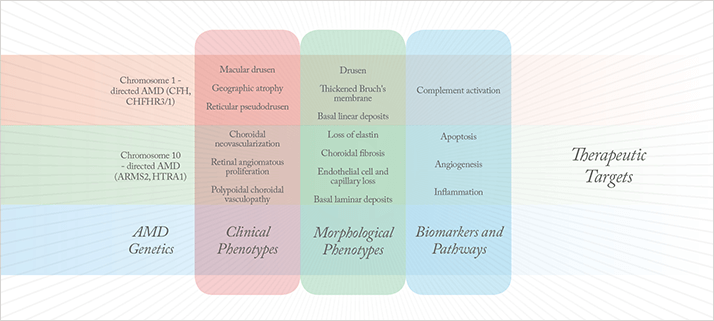
Aberrant function of the complement system at the level of the retinal pigment epithelium (RPE)-choroid interface plays a key role in AMD disease etiology. This was discovered in early morphological and immunohistochemical studies of human donor eyes. Subsequent genetic analyses identified significant associations with a number of complement pathway-associated genes: a specific common haplotype of the complement factor H (CFH) gene on chromosome 1, which accounts for a significant proportion of risk for developing AMD (1-5); a pair of common protective haplotypes (6-8), one of which – a large deletion distal to CFH – encompasses the CFH-related 1 (CFHR1) and CFHR3 genes; and variations in other complement regulators, including complement factor B (CFB) (9) and complement component 3 (C3) (10). Taken together, these complement-related variations account for a major proportion of the genetic risk for AMD in the Caucasian population. Importantly, variants within a locus containing the non-complement pathway-associated ARMS2 and HTRA1 genes on chromosome 10 have a strong and significant association with AMD risk (11-13). It has not yet been possible to assign frank AMD causality to either of these two genes. Moreover, they certainly have no obvious, documented relationship to the complement pathway. HtrA1 is a serine protease known to participate directly or indirectly in the degradation of extracellular matrix (14-16). It is intriguing to speculate that HtrA1 could participate in the development of late-stage AMD via the degradation of Bruch’s membrane, an event that typically precedes choroidal neovascular membrane formation. Little is known about the function(s) of the ARMS2 gene; indeed, strong evidence that the protein is actually synthesized is lacking.
Genotype-Phenotype Associations
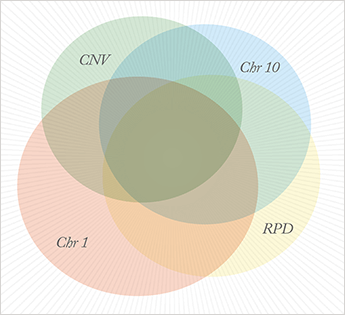
In combination, the AMD-associated genes on chromosomes 1 and 10 account for a majority of disease risk. Although AMD is typically described by most investigators as a single, complex-trait disease, our analyses indicate that AMD may instead represent multiple, distinct biological diseases that exhibit significant overlap within the population. Our general approach has been to analyze genetic ‘outliers’, that is, cases who are homozygous risk at chromosome 1, but without any risk alleles at chromosome 10, which we term ‘chromosome 1-directed AMD’; and cases who are homozygous risk at chromosome 10 without any risk alleles at chromosome 1, which we call ‘chromosome 10-directed AMD’. We have found that the two major advanced forms of AMD, namely geographic atrophy (GA) and choroidal neovascular disease (CNV), associate independently with both chromosome 1- and chromosome 10-directed AMD, both clinically and histologically. In contrast, macular drusen (the traditional clinical ‘marker’ for early stage AMD) are significantly associated with chromosome 1-directed, but not with chromosome 10-directed, AMD. This suggests that drusen formation is far more robust, and likely driven by the chromosome 1 locus, a suggestion supported by the observations that the development of CNV is not preceded by the deposition of macular drusen in 8-10 percent of patients with CNV and that a majority of these patients carry increased genetic risk at chromosome 10. Moreover, our preliminary evidence suggests that ocular histological phenotypes in individuals with chromosome 1- and chromosome 10-directed disease differ markedly and that the biology and response to anti-VEGF drugs differs markedly between patients with chromosome 1- and chromosome 10-directed disease.Additional evidence for the ‘multiple disease model’ comes from our recent pilot serum biomarker studies conducted on chromosome 1- and chromosome 10-directed AMD patients. Using a custom panel of protein biomarkers, we have identified a number of specific associations with either chromosome 1 or chromosome 10 risk status.
Reticular Pseudodrusen Phenotype
Another focus in our laboratory over the past few years has been the characterization of a distinct and newly-recognized phenotype of AMD, termed ‘reticular pseudodrusen’ (RPD; 17-19). RPD represents a distinct phenotype that overlaps substantially with those of chromosome 1- and chromosome 10-directed phenotypes. We have learned that the formation of RPD precedes, and is predictive of, 65-70 percent of the severe, late-stage forms of AMD in our cohorts. Although the precise nature of RPD is debated, it is clear that infrared-detectable RPD lesions are largely restricted to the macula (within the vascular arcade) and that individual RPD lesions average 200–250 ?m in diameter. The sizes and distribution of these lesions are regular and we have proposed that they recapitulate the topology of macular choriocapillaris lobules, suggesting the possible involvement of the choriocapillaris and/or choroidal vasculature in their origin, or that these structures become more visible to infrared imaging in various disease states. Genetically, RPD are more strongly associated with chromosome 10 than chromosome 1, although neither chromosome is as strongly associated with RPD as they are with choroidal neoavascularization. Importantly, RPD are also associated with the development of late-stage AMD in the absence of chromosome 1 or 10 risk alleles. These observations suggest that either RPD manifest as a result of an as-yet-undiscovered AMD-associated gene(s) or that they represent a specific stage of retinal degeneration that is common to chromosome 1- and chromosome 10-associated disease. Resolving this issue is imperative.Research Resources and Strategies
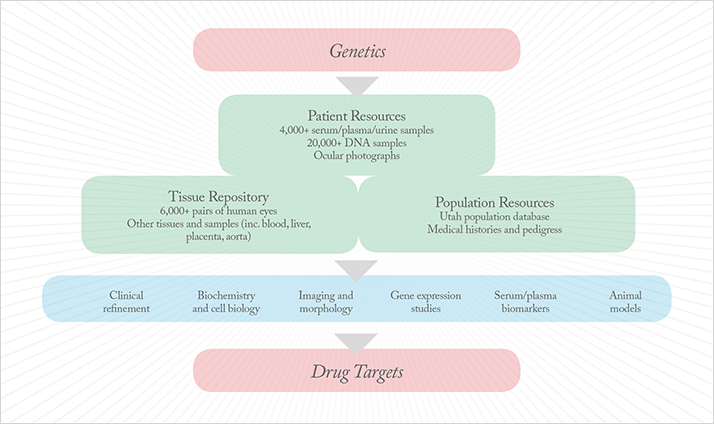
Our team now believes that there is ample available evidence to suggest that AMD comprises multiple, distinct biological diseases that exhibit significant overlap within the population (see Figure 2). However, more extensive studies will be required to validate it. Right or wrong, it is my hope that this concept will serve to drive the field to explore this hypothesis, to ask more rigorous questions, and to come to an even deeper understanding of the biology underpinning this devastating condition. Our current studies are designed to further illuminate the clinical phenotypes, ultrastructural morphology, and biology of RPD-, chromosome 1-, and chromosome 10-directed disease and to test our concept that AMD is distinct biological diseases. I anticipate that these studies will ultimately provide a comprehensive understanding of the role of risk conferred by RPD, chromosome 1, and chromosome 10 on AMD pathology. One key resource in this research is human eyes donated at the time of death to study pathways associated with the development of AMD. My colleagues and I first conceived of this approach in 1987, while I was a faculty member at the Bethesda/Anheuser Busch Eye Institute in Saint Louis. The guiding concept was a simple one: acquire sufficient numbers of eyes from donors with and without previous clinical histories of AMD, such that rigorous biological assessment of these two groups of tissues might reveal pathways that are associated specifically with AMD. This approach has remained a pivotal component of our research program to this day, while technological advances during the past 20 years have made it far more powerful.
Our donor eye repository is now comprised of eyes and other tissues from over 6,500 well-characterized donors. It is the world’s largest repository of donated human eyes; in fact, the next largest has just a few hundred eyes. The vast majority of eyes are processed within four hours of death. Medical and ocular histories, a family questionnaire, blood and sera have been obtained from every donor and we maintain a database of ophthalmological, medical and scientific information related to each one. In total, we have verified clinical histories of AMD for 24 percent of the eyes in the collection, and numerous other well-documented ophthalmic and systemic diseases are also represented. Over 1,200 pairs of eyes have been donated to the Moran Center for Translational Research (CTM) repository during the past three years alone and we have launched an expanded effort to recruit currently enrolled study patients to become eye donors upon their deaths. Of these donors, 22 percent were patients seen previously at the John Moran Eye Center, thus providing CTM staff with the unique capability to establish genotype-phenotype associations. From these, DNA, RNA, protein, fixed tissue, and frozen tissues are prepared for experimental analysis. Data derived from these tissues and from our patient case-control cohorts are playing a critical role in the identification of key AMD-associated genes, pathways and therapeutic targets.
I will always be extremely grateful to all the individuals and families for their precious gifts of these eyes. Having spent thousands of hours with the families who make the decision to donate, I have learned how unselfish these gifts are; I will always be a respectful steward of thisvaluable resource. The CTM cohort of AMD case and control subjects is currently comprised of greater than 5,000 Utah study patients with and without AMD, and an additional 10,000 study patients from other locations, including cohorts with a variety of ethnic backgrounds. Medical, ophthalmological and imaging data, as well as DNA, serum, plasma, and urine is available for most of these study patients; these resources are critical for coordinated genomic, proteomic, epidemiologic and other studies. Our recruitment of study subjects is ongoing and is focused on refinement of the existing cohorts and the addition of subjects with the RPD phenotype. We currently have DNA samples from over 1,000 individuals with RPD in the collection. We are concentrating our efforts on patients with ‘pure’ chromosome 1 and chromosome 10 genotypes, especially those rarer individuals homozygous for chromosome 10 risk. These patients are undergoing clinical evaluation of disease progression, as well as the collection of additional serum, plasma, and white blood cell samples that will be employed in the protein biomarker and gene expression studies.
Ongoing research is a continuation of our integrated effort to refine genotype-phenotype associations in AMD, with the long-range goal of identifying biological pathways that are specifically associated with these various gene-driven phenotypes. This multidisciplinary research program is designed to elucidate further chromosome 1-, chromosome 10- and RPD-directed pathways, and to identify therapeutic targets that will form the basis of future drug development efforts. Specific ongoing and future research activities include:
- Identification of diseases that co-segregate with specific ‘forms’ of AMD.
- Characterization of disease progression in donor eyes based upon disease-specific markers identified through gene expression studies.
- Genetic analysis of the RPD phenotype, using genome-wide association studies (GWAS) and/or whole exome sequencing of our extensive case-control cohort.
- Genetic and phenotypic analyses of non-chromosome 1 and non-chromosome 10 AMD.
- Identification of the causal chromosome 10 gene through the directed characterization of ARMS2 function and HtrA1 expression in AMD samples.
This thorough characterization of the biology of specific clinical and genetic patient populations will play a critical role in refining our understanding of the pathogenesis of AMD in its various forms. It will also greatly assist efforts to develop disease-directed diagnostics, to ascertain and stratify patients for future clinical trails, and to identify drug targets and to develop effective therapeutic modalities for this devastating disease. Selecting a pharmaceutical partner to collaborate with us is an important component of our development plan and we are currently in discussions with a number of companies interested in forming a partnership. We want to work side-by-side with our partner to identify and validate gene-directed targets, potentially shortening the drug development delivery time by years. The Moran Center for Translational Medicine – a close family of clinicians, scientists, technicians, patients, donors, and hundreds of other supporters who have joined the fight for better vision care and medicine – is up to the challenge of developing new treatments for this devastating condition. I have little doubt but that the ultimate goal of halting or delaying the progression of this blinding disease will be accomplished. We will find a cure for AMD in my lifetime – all of us, working together. Gregory Hageman is the John A. Moran Presidential Professor, Department of Ophthalmology and Visual Sciences, and Director, Moran Center for Translational Research in Salt Lake City, Utah, USA.
References
- A. O. Edwards, R. Ritter III, K. J. Abel, et al., “Complement factor H polymorphism and age-related macular degeneration”, Science, 308, 421–4 (2005). G. S. Hageman, D. H. Anderson, L. V. Johnson, et al., “A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposesindividuals to age-related macular degeneration”, Proc. Natl. Acad. Sci. USA, 102, 7227–32 (2005). J. L. Haines, M. A. Hauser, S. Schmidt, et al., “Complement factor H variant increases the risk of age-related macular degeneration”, Science, 308, 419–21 (2005). R. J. Klein, C. Zeiss, E. Y. Chew, et al., “Complement factor H polymorphism in age- related macular degeneration”, Science, 308, 385–9 (2005). S. Zareparsi, K. E. Branham, M. Li, et al., “Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration”, Am. J. Hum. Genet., 77, 149–53 (2005). G. S. Hageman, L. S. Hancox, A. J. Taiber, et al., “Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications”, Ann. Med., 38, 592–604 (2006). A. E. Hughes, N. Orr, H. Esfandiary, et al.,, “A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration”, Nat. Genet., 38, 1173–7 (2006).? S. Raychaudhuri, S. Ripke, M. Li, et al., “Associations of CFHR1-CFHR3 deletion and a CFH SNP to age-related macular degeneration are not independent”, Nat. Genet., 42, 553–5 (2010). B. Gold, J. E. Merriam, J. Zernant, et al., “Variation in factor ?B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration”, Nat. Genet., 38, 458–62 (2006). J. R. Yates, T. Sepp, B. K. Matharu, et al., “Genetic Factors in AMD Study Group. Complement C3 variant and the risk of age-related macular degeneration”, N. Engl. J. Med., 357, 553–61 (2007). J. Jakobsdottir, Y. P. Conley, D. E. Weeks, et al., “Susceptibility genes for age-related maculopathy on chromosome 10q26”, Am. J. Hum Genet., 77, 389–407 (2005). A. Rivera, S. A. Fisher, L. G. Fritsche, et al.,, “Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk”, Hum. Mol. Genet., 14, 3227–36 (2005). Z. Yang, N. J. Camp, H. Sun, et al., “A variant of the? HTRA1 gene increases susceptibility to age-related macular degeneration”, Science 314, 992–992 (2006). E. An, S. Sen, S. K. Park, et al., “Identification of novel substrates for the serine protease HTRA1 in the human RPE secretome”, Invest. Ophthalmol. Vis. Sci., 3379–3386 (2010). S .Grau, P. J. Richards, B. Kerr, et al., “The role of human HtrA1 in arthritic disease”, J. Biol. Chem., 281, 6124–6129 (2006). A. Chamberland, E. Wang, A. R. Jones, et al., “Identification of a novelHtrA1-susceptible cleavage site in human aggrecan: evidence for the involvement of HtrA1 in aggrecan proteolysis in vivo”, J. Biol. Chem., 284, 27352–27359 (2009). J. J. Arnold, S. H. Sarks, M. C. Killingsworth, J. P. Sarks, “Reticular pseudodrusen. A risk factor in age-related maculopathy”, Retina, 15, 183–91 (1995). S. Y. Cohen, L. Dubois, R. Tadayoni, et al., “Prevalence of reticular pseudodrusen in age-related macular degeneration with newly diagnosed choroidal neovascularization”, Br. J. Ophthalmol., 91, 354–9 (2007). S. A. Zweifel, Y. Imamura, T. C. Spaide, et al., “Prevalence and significance of subretinal drusenoid deposits (reticular pseudodrusen) in age-related macular degeneration”, Ophthalmol., 117, 1775–81 (2010).
