In our last installment we heard from BBC journalist Ramadan Younes about his experience with retinitis pigmentosa and his investigation detailing some of the fraudulent treatments for this condition. To gain deeper insight into the etiology of RP, how it can be managed and what the promising avenues for a future treatment may be, we spoke with Power List member Mariya Moosajee who is Professor of Molecular Ophthalmology and Consultant Ophthalmologist at Moorfields Eye Hospital and leading expert on the genetics of inherited retinal diseases.
Could you please walk us through what we know about RP’s etiology and how ophthalmologists typically screen and diagnose RP and other inherited retinal diseases?
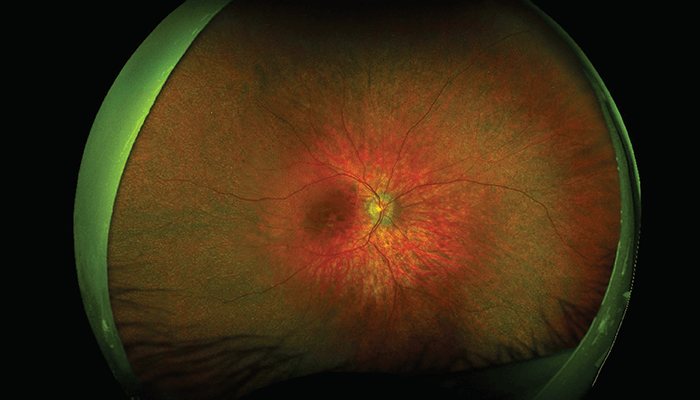
RP is the most common inherited retinal disease, characterized by progressive retinal degeneration with initial impairment of rod photoreceptors, resulting in nyctalopia and peripheral visual field loss, which can begin in childhood or later in adulthood. This initial degeneration is followed by cone involvement and eventual central vision loss. Patients may also experience photopsia, photophobia, and dyschromatopsia.
Examination of the fundus typically reveals optic disc pallor, retinal vessel attenuation, and intra-retinal bone-spicule pigmentation in the mid-periphery, with varying degrees of retinal pigment epithelium (RPE) atrophy. In early stages, you may only see an abnormal/impaired foveal reflex or subtle RPE mottling/areas of depigmentation. However, fundus autofluorescence will reveal a central hyperautofluorescent ring that constricts over time.
In 20–30 percent of patients, RP is syndromic with systemic manifestations – the most common being Usher syndrome and Bardet-Biedl syndrome, hence it is important to undertake a systems review.
RP is a genetic eye disease caused by mutations in over 80 genes, which can be inherited in an autosomal recessive (AR), autosomal dominant (AD) and X-linked manner. It is now imperative that we undertake genetic testing on patients so we can identify the molecular cause of their RP. Such testing allows us to offer the patient and their family accurate genetic counseling, family planning advice, and access to pertinent research, clinical trials, and any potential treatments that may be available.
There is no cure – but what management options are currently available?
As Ramadan noted in Part I, there is an approved gene therapy (voretigene neparvovec) available for patients with biallelic RPE65 mutations, which can cause a rare form of RP. For most, supportive management is required. Patients can develop refractive errors, early-onset cataracts (typically posterior subcapsular lens opacities), cystoid macular edema (up to 50 percent of patients may be affected), macular holes, and epiretinal membranes.
Annual follow-up is vital to assess for such complications and provide the appropriate interventions. It is also key to offer referral to low vision services and access to assistive digital technology – and to consider sight impairment registration. Lifestyle advice includes encouraging a healthy diet consisting of fresh fruit and green leafy vegetables, regular exercise, and UV protected sunglasses.
What are the most promising avenues of research? And what do you hope we’ll achieve in the next 10 years or so?
Gene therapy using adeno-associated viral (AAV) vectors for other genetic causes of RP is underway at clinical trial stage. Alternative strategies for gene augmentation are imperative due to emerging drug-induced chorioretinal atrophy seen in patients treated with voretigene neparvovec (1). Non-viral gene therapy using DNA plasmid vectors can accommodate genes of any size and lacks any viral components so should be safer for patients, whilst allowing for repeated administration, if required.
RNA therapy includes antisense oligonucleotides (AON), which are short strands of nucleotides that bind to their target messenger RNAs (mRNA) to block the transcription of a mutant allele or correcting a splicing defect at the pre-mRNA level. These are delivered through intravitreal injection and may play a role in future treatments for IRDs.
Gene editing through the CRISPR/Cas9 system can repair genetic mutations by acting like a custom pair of scissors programmed to cut out an abnormal section of a gene and introduce the correct sequence of letters.
Optogenetics aims to restore some level of vision in patients with advanced RP by conferring photo-detection abilities to the surviving inner retinal cells. This is achieved by transfecting cells with genes encoding for light-sensitive ion channels or pumps, such as channelrhodopsin (ChR) and halorhodopsin, which are derived from microorganisms.
AAV vectors can also be used to deliver certain factors back to the retina in a more agnostic approach; for example, rod-derived cone viability factor, which preserves the integrity of cones. Other oral drug treatments as a form of neuroprotection are also being investigated; for example, N-acetylcysteine (NAC)/N-acetylcysteine-amide (NACA) is a high dose antioxidant that may be able to reduce oxidative stress and ensuing photoreceptor damage.
Cell replacement therapy using human retinal progenitor cells is also underway. Here, the theory is that these cells may release neurotrophic factors to protect the surviving photoreceptors. A subretinal delivery method has reached phase III clinical trials.
In the next 10 years, I would like to see many more effective therapies approved for use in our patients – but only once they have been investigated rigorously to ensure there are limited safety issues. I hope that these new therapies can halt or at least slow disease progression and that there are options for patients at various stages of their disease, even combination therapies. Most importantly, I want the global approach to be patient-centered; for example, sharing resources to reduce duplication in research and waste of funds, ensuring open access to results so we can learn from pitfalls and successes, and using greater collaboration to access individual strengths and expertise to advance projects through clinical translation.
What are the pseudo-treatments on offer – and what are the risks?
These rogue treatments are incredibly worrying and I have spent most of my career advising patients against such endeavors. These pseudo-therapies have no proven evidence-base for their effectiveness in treating these conditions. I worked in the UAE and was shocked by the cases I saw, many had spent a fortune traveling to centers claiming their “treatment” would restore their vision. I had small children undergoing operations to increase the blood flow to the retina by receiving incisions in the region of the temporal artery and vein; they are now left permanently scarred. Some patients had leeches placed on their eyes to draw out bad blood. Many had stem cell injections from clinics all over the world – intraocular, retrobulbar, sub-tenons, and intravenous injections. None reported any improvements, and in some they had lost vision acutely or permanently.
There are huge risks to patients, mainly in terms of the fact that they do not know what treatment they are consenting too (especially if in another language), they do not know what is being administered to them and how, and they do not understand the risks, such as bleeding, infection, scarring, inflammation, and further sight loss.
Some patients are told there are no risks, but all procedures and drugs have some risk or at least a significant list of potential side effects/complications that can arise.
And what do you think about the “practitioners” pushing these bogus therapies?
I am wholly disappointed and angered by any practitioner who advocates for these therapies. They need to stop and consider whether they would want the same for themselves or their own family. The sight loss community is shocked and dismayed by these operators. If they are truly qualified ophthalmologists, with permission to practice, they must integrate into the scientific and clinical IRD community and understand the research and development processes; for example, how to set up and fund clinical trials ethically. They must learn and understand the principles of good medical and research practice – and gain some continued medical education in the field. The authorities in each respective country need to have stricter laws to govern this unlawful activity and always place patient safety at the heart of their practice.
What needs to change in terms of regulation and awareness?
It is very hard to comment on the regulations and laws outside of the UK. Patients are relatively well protected in the UK and more so within the NHS. But in the private sector, this can be very different. One key example is the case of ophthalmologist Bobby Qureshi – the director of the London Eye Hospital who was offering telescopic intraocular lens implant surgery for AMD and charging £24,000. The Macular Society, a leading UK sight loss charity focused on macular disease, submitted a complaint to the GMC in 2017 after receiving more than 50 telephone calls complaining about Qureshi. The Society was concerned that Qureshi had cheated patients and was operating on people inappropriately. He was struck off the GMC register in 2019. (2)
There is no portal or international governing body that can regulate practitioners from these activities across the world, but we can strive to educate patients and empower sight loss charities to disseminate information and raise awareness within their communities to hopefully prevent exploitation.
All sight loss charities need to be aware of these bogus treatments and be able to advise patients on whether a potential trial is worth pursuing or not. Or to sign post individuals to genetic eye specialists, who can help. They also need to be on guard if they receive reports from their members about sight threatening outcomes from trials so they can represent the community and raise concerns on their behalf.
I created the website Gene Vision as a reliable A to Z of genetic eye disease for patients and professionals so they can learn more about rare genetic eye diseases (3). I include links to the management, support groups/charities, and latest research. We have over 3000 new users per week and individuals from over 200 countries (USA, UK, and India being the top users) access the site. But more work needs to be done to ensure this information can be accessed by patients worldwide; for example, translating the content into different languages and adding links and processes that may be more country specific.
What can ophthalmologists learn from the documentary – and how can the global ophthalmology community help protect patients from unscrupulous actors?
From my perspective, the global IRD community is relatively well connected and we are aware of those specializing in the field and their work – aided by attending international scientific meetings, such as ARVO, AAO, Euretina, and so on, where researchers can present their work and the results. Publishing outcomes of the research in peer-reviewed scientific journals also helps us understand whether there is any evidence base for emerging therapies. The problem is that those offering rogue treatments are often not necessarily engaging in good governance. Potentially, we need to form an international committee with a portal that allows for the reporting of unethical activity and can be accessed by patients around the world.
What should patients be aware of when looking at clinical trial information?
I can share a few warning signs: If there is no public record of peer-reviewed preclinical research by the investigators; if participants are charged to participate; if the treatments are unapproved by the US Food and Drug Administration or the European Medicines Authority; if the researchers do not mask investigators or participants to the intervention; if they do not randomize patients; if they do not have a control group of placebo or sham procedure.
We often use ClinicalTrials.gov – a database provided by the US National Library of Medicine – to identify new studies on RP, and it is a very useful resource; however, its weakness is that it can be used to advertise “pseudo-studies” like the SCOTS2 trial highlighted in the documentary (4).
If you are interested in a particular trial, it’s important that you contact your doctor and ask their advice on whether they think it is reputable and ethically approved.
Finally, please watch this documentary and share with others.
References
- F. Reichel et al., “Development of retinal atrophy after subretinal gene therapy with voretigene neparvovec,” Br J Ophthalmol, (2022) Online ahead of print. PMID: 35609955 and W.S Gange et al., “Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis,” Ophthalmol Retina, 6, 58, (2022) PMID: 33838313.
- Macula Society, “High profile eye surgeon struck off,” available at: https://bit.ly/3LsMyTh
- Gene Vision, “Retinitis Pigmentosa for Patients,” available at: https://bit.ly/43UQDa9
- See the SCOTS2 trial entry at the US National Library of Medicine: https://bit.ly/43YFUvl
