
By Geoffrey Potjewyd after interviewing Eva M. del Amo Páez, Adjunct Professor at University of Eastern Finland, Kuopio, Finland.
Ocular diseases are an increasing societal problem, exacerbated by our aging population, environmental factors, and a global epidemic of diabetes. The lack of effective treatments for more than a handful of diseases is a compounding problem. Industry and academia are, of course, both keen to advance ocular drug discovery and development in these therapeutic areas, but do we have the right tools at our disposal?
When developing or administering any therapeutic, we must understand how the drug will be affected by the body – the pharmacokinetics. Pharmacokinetics includes drug absorption, distribution, and elimination. The latter may be via metabolism (by changing the drug) or excretion out of the body. Unfortunately, metabolism of ocular drugs are often assumed from data on what occurs in organs far removed from the eyes.
Indeed, information on drug-metabolizing enzymes within the eye is sparse, and I believe new insights in this field would benefit the current and future development of ocular therapies. And that’s why it’s a major research theme for my team at the University of Eastern Finland, Kuopio, Finland.
Drug metabolism
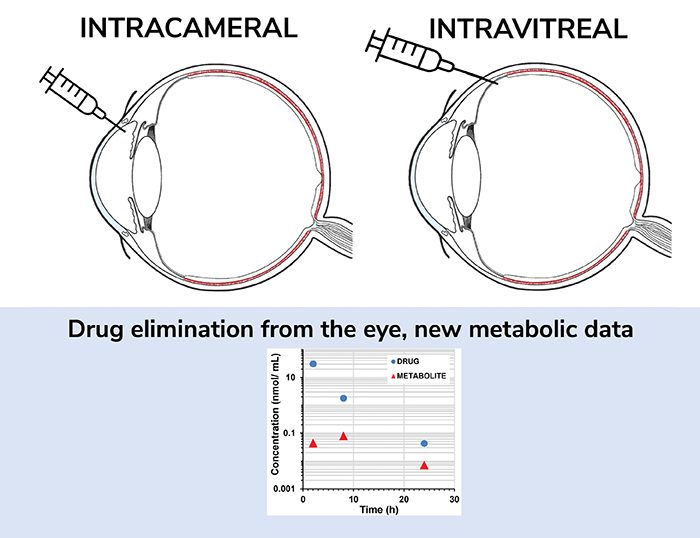
In fact, we directly address the lack of information on ocular drug-metabolism in our recent publication (1), where we present a comprehensive ocular pharmacokinetic study that investigates the metabolism and distribution of four drugs in the rabbit eye. Each drug – acetaminophen, brimonidine, cefuroxime axetil, and sunitinib – was selected because it interacts and is broken down by distinct enzymes (sulfotransferase, aldehyde oxidase, esterases, and CYP3A, respectively). The drugs were applied through either intracameral or intravitreal routes, and concentrations of both injected drug and the main metabolite of the drug, were measured in six different ocular tissues to determine the route taken. Pharmacokinetic drug and metabolite profiles were then obtained and analyzed.
Interestingly, extremely low levels of metabolites were detected for all the compounds, except the esterase substrate, which shows that drugs are mostly eliminated from the eye by non-metabolic processes, but via “excretory” action of the ocular flows (i.e. metabolism played a minor role in their ocular pharmacokinetics). In simple terms, our study shows that you cannot reliably predict ocular metabolism from data generated through tests for hepatic metabolism (the data that are generally produced during drug development). Only esterases seem to have a major impact on ocular drug clearance.
Ocular impact
As noted, the main mechanism of ocular drug elimination is not metabolism-related, but ocular flow-related (how well the drug can permeate the ocular membranes and be cleared by aqueous humor and ocular blood flows). Typically, intravitreal small molecular weight drugs, such as the ones we investigated, are cleared from the eye rapidly, so controlled drug delivery systems are essential to prolong their therapeutic action in the eye (such as the Ozurdex intravitreal implant containing dexamethasone). For future drug delivery systems, we can expect that pharmacokinetics will not be controlled by drug metabolism. Nevertheless, some very low-level of metabolite formation is expected, and the potential ocular toxicity from the metabolite should be evaluated during drug design and development, especially for these long-acting formulations.
In clinics, antibiotics and antiviral drugs are intravitreally injected to treat endophthalmitis and viral infections (such as cytomegalovirus infections) respectively, while intracameral injections of antibiotics may be given after cataract surgery. Based on our findings, ocular-metabolism will not increase the clearance of these drugs, with the decrease of drug half-life (again, esterase substrates are the exception).
This finding is in line with our previous research, where we observed drug hydrophilicity (chemical affinity to water) to be the key parameter for prolonging duration of ocular residence for intravitreal drugs (2). Hydrophilic drugs permeate less through the blood-ocular barriers and, therefore, have reduced passage into systemic circulation of vascularized tissues. In other words, intravitreal hydrophilic drugs present slower clearance and longer half-lives in the vitreous.
When attempting to select an antibiotic from two with equal potency, we suggest that physicians choose the most hydrophilic one to prolong the effect of the treatment and reduce the number of injections for the patient. Tip: hydrophilicity is easily obtained from web databases, such as drugbank.com, by looking for the one with lowest logD7.4 or logP values.
Good Model
In our research, we used a cassette-based dosing methodology, which included the four drugs in the same injection solution. And we used a rabbit model, which has been proven as a good model for ophthalmic intravitreal drugs (3). The combination of these two factors means that we were able to reduce the use of the number of animals according to the principle of 3Rs (reduce, refine, replace), with the added bonus of diminishing variability between studies. In future, when investigating a new intravitreal drug, where preclinical pharmacokinetic studies are compulsory, I would definitely choose the rabbit model for its relevance and accuracy.
Diving into the data
Our pharmacokinetic data in six different ocular tissues for four compounds is rich and informative, depicting four clearly different drug behaviors in the eye. The modeling of these drug concentrations in the different compartments should provide a better understanding of the intravitreal and intracameral pharmacokinetic processes specifically for these compounds, and in general on small molecular weight drug distribution and its relationship with physicochemical properties.
The parameters established in our work could also be used for pharmacokinetic simulations to guide the design of new ophthalmic drug delivery systems, informing on the relationships between drug release rates and loading doses of the systems for a specific duration of therapeutic action. They could also feed into simulations for other routes, such as topical, subconjunctival, intravitreal, and intracameral administration to predict drug concentrations in the anterior and posterior segment of the eye. We believe that further modeling on this data will give more insights into the pharmacokinetics and distribution of these drugs.
Another interesting avenue of research would be to develop rabbit-based models of choroidal or retinal neovascularization to investigate the effect of the drug on the disease state while exploring drug pharmacokinetics.
Drug developers: over to you!
 Eva del Amo Páez is the Adjunct Professor at University of Eastern Finland, Kuopio, Finland.
Eva del Amo Páez is the Adjunct Professor at University of Eastern Finland, Kuopio, Finland.
References
- EM Del Amo et al., “Ocular metabolism and distribution of drugs in the rabbit eye: Quantitative assessment after intracameral and intravitreal administrations,” Int J Pharm, 613, 121361 (2022). PMID: 34896561.
- EM Del Amo, et al., “Intravitreal clearance and volume of distribution of compounds in rabbits: In silico prediction and pharmacokinetic simulations for drug development,” 95, 215 (2015).PMID: 25603198.
- EM Del Amo, A Urtti, “Rabbit as an animal model for intravitreal pharmacokinetics: Clinical predictability and quality of the published data,” Exp Eye Res, 137, 111 (2015). PMID: 25975234.
