- Anti-VEGF agents have transformed our ability to treat wet AMD – but this is essentially late-stage, palliative care; we still have no treatments for geographic atrophy or earlier interventions beyond nutritional supplements.
- A better understanding of AMD’s pathogenesis – and its genetic underpinnings – is helping us understand what needs to be addressed by novel therapies.
- A “big data” approach is helping us understand the interaction between major epidemiological risk factors for developing AMD, and their relationship to phenotype.
- Clinical and basic research is often performed in splendid isolation. Only by working together can we bring together the knowledge required to deliver new therapies for AMD.
Ὁ βίος βραχύς, ἡ δὲ τέχνη μακρή, ὁ δὲ καιρὸς ὀξύς, ἡ δὲ πεῖρα σφαλερή, ἡ δὲ κρίσις χαλεπή.
Life is short, and art long, opportunity fleeting, experience perilous, and decision difficult.
Aphorismi (Hippocrates)
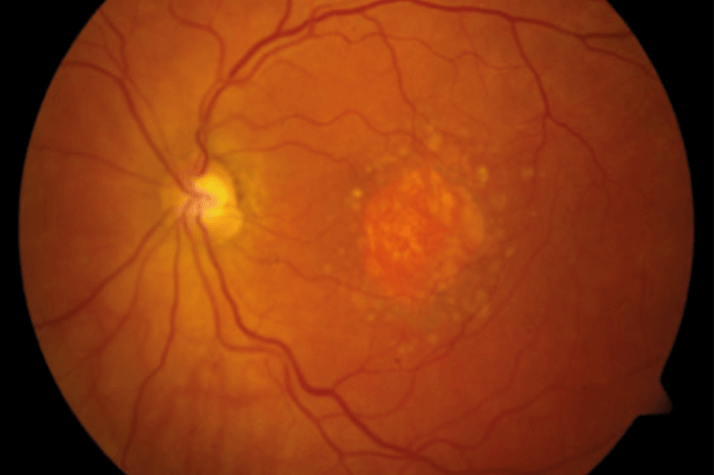
In Ancient Greece, life was indeed short in comparison with the enduring art of medicine. However, longevity presents new challenges. For many of us, life is now long – perhaps too long for our eyes. Owing to a combination of genetic and environmental factors, people now often outlive their maculae (Figure 1). Despite anti-VEGF drugs, age-related macular degeneration (AMD) remains the leading cause of blindness in developed countries. Indeed, our aging population means that many thousands of people will continue to lose their vision and independence unless we can develop new treatments that target the underlying disease processes in AMD.
Opportunity is fleeting
While the anti-VEGF era has seen a tremendous advance in our approach to AMD, the window of opportunity for initiating this therapy is very short. In some senses, anti-VEGF therapy is palliative medicine. We observe patients until they have the most advanced form of AMD before injecting an eye with drugs that fail to target the underlying disease process. Significant tissue damage and visual loss may have already taken place and, further, some patients respond poorly to these treatments. We still have no treatments in routine use for geographic atrophy, which is thought to affect over 8 million people worldwide. Ideally, future treatments should target the underlying disease process, preferably during early-stage AMD, and be personalized to patient genotype. To make this a reality, we need a refined understanding of AMD pathogenesis, and specifically one that is predicated on our knowledge of its genetic basis. The progress in our grasp of AMD genetics has been a fantastic success story over the past decade. It is now 10 years since four papers reported a strong association between the Y402H polymorphism in the Complement Factor H (CFH) gene on chromosome 1; a second strong association with a locus in ARMS2/HTRA1 on chromosome 10 was demonstrated soon after. However, we have perhaps been slow to discover the precise biochemical mechanisms of disease related to each of these loci.Following clinical training in ophthalmology and a PhD studying the biochemistry of aging changes in the human macula, I spent a year as a Fulbright Fight for Sight Scholar doing research at the Center for Translational Medicine in the Moran Eye Center (University of Utah). This work was with Gregory Hageman, who has been a pioneer in demonstrating that AMD may represent at least two partially distinct biological diseases: one driven by the complement system through risk at chromosome 1 (the CFH locus) and the other associated with risk at chromosome 10 (the ARMS2/HTRA1 locus). Many of our patients with AMD will have risk variants at both these loci but studying those individuals with risk at only one locus or the other has been pivotal in examining these subtypes of AMD in their purest forms. The main message is that patients with ‘pure 1 disease’ have a greater tendency towards formation of large drusen in the central macula and development of geographic atrophy, though they do also develop neovascular disease; patients with ‘pure 10 disease’, by contrast, exhibit relatively few macular drusen but are strongly predisposed towards neovascular disease, with a high incidence of retinal angiomatous proliferative (RAP) lesions (1). Understanding AMD in this way has important implications. It helps explain some marked geographical differences in disease phenotypes and appearance, since chromosome 1 disease is more common in Caucasian populations and chromosome 10 disease more prevalent in Asian populations. It also means that we have two sets of biological pathways pathogenesis fully. Most importantly, it means that we will need at least two, genotype-dependent, sets of therapies for patients with AMD. In the interim, it certainly means that we need to select patients very carefully (on the basis of genotype as well as phenotype) for inclusion in clinical trials, depending on whether the drugs being evaluated target AMD related to chromosome 1 or chromosome 10.
Experience is perilous
The use of animal models in an attempt to replicate AMD has significant limitations and may mislead. In addition, the complement system is notoriously difficult to study in vitro. For a truer understanding of the disease, it is essential to study genetic and biochemical changes at the site of disease formation in the species of interest. I therefore feel that it is vital to use human ocular tissue in order to understand AMD pathogenesis. I have been lucky enough to work at the Moran Eye Center and the University of Manchester, both of which have superb access to large banks of human macular tissue. In fact, through the generosity of many donors (including those with AMD), Gregory Hageman has, over the past 20 years, been able to create a repository of over 7,000 pairs of human eyes – the largest collection anywhere in the world. Crucially, each pair of eyes has extensive accompanying information about the donor, including AMD genotype, medical history, and even retinal imaging carried out clinically over many years prior to death. At the Moran Eye Center, under the auspices of Gregory Hageman, I was able to perform the first published study of pure chromosome 1-directed AMD using human macular tissue (2). We demonstrated conclusively that, even before clinical AMD develops, individuals with genetic risk at chromosome 1 have significantly higher levels of complement activation at the macular RPE-choroid interface. We also showed that cigarette smokers have significantly increased complement activation, oxidative stress and inflammation at the same site, compared to non-smokers of the same genotype (Figure 2). Clearly, the mixture of genetic predisposition and smoking is a potent combination that over decades can prove too toxic for the vulnerable human macula. This work provides a compelling rationale for complement inhibitors in reducing AMD progression, particularly for geographic atrophy. Most importantly, it shows that the best candidates for clinical trials of these drugs will be patients with chromosome 1 disease.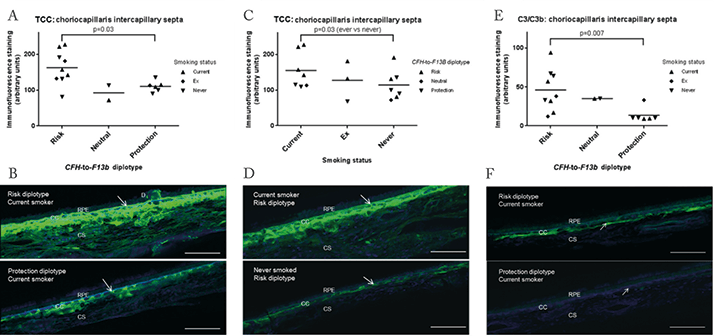
A second vital question to address is how exactly genetic risk at chromosome 1 leads to increased complement activation. Funded by Fight for Sight, I spent my PhD years at the University of Manchester examining this question, together with Paul Bishop, Anthony Day, and Simon Clark. In fact, we discovered a new potential disease mechanism for AMD (3). In Bruch’s membrane, CFH is required in order to prevent excessive complement activation, which leads to inflammatory damage. In the human macula, CFH relies on particular sugar chains called heparan sulfate to bind to Bruch’s membrane. Using human macular tissue, we made two very important observations: first, that the risk variant of the CFH protein binds poorly to heparan sulfate (4) and, second, that levels of heparan sulfate in human macular Bruch’s membrane decline very substantially with age (3). This has very significant implications and may help explain why AMD takes decades to develop. It means that chromosome 1-directed AMD develops through a double hit of genetic risk at CFH together with age-related changes in the human macula, leading to excessive complement activation and a downwards spiral to inflammation, macular damage and visual loss.
A ‘big data’ approach
Over the past decade, I have also worked closely with the professor of public health at the University of Oxford, Michael Goldacre, at the institution’s Unit of Health-Care Epidemiology. Our approach has been to bring ‘big data’ to bear on important questions related to AMD. We have used routinely collected data – information collected on all patients treated in the English National Health Service – and have published multiple studies, most of which have been the largest and longest studies of their kind in AMD research. Importantly, we have used record linkage with these datasets in order to trace individuals through multiple diagnoses over long time periods – an extremely powerful tool for association studies. For example, we analyzed an AMD cohort of 66,000 people over a decade to demonstrate that neovascular AMD and Alzheimer’s disease are not associated in the English population (5). Our biggest study group so far has been an English osteoarthritis cohort of over 2 million people. We followed these individuals for over a decade and found that people with arthritis have a modestly increased risk of neovascular AMD (6). These studies were conducted on individuals with AMD associated with any genotype; the next step would be to perform association studies on groups of AMD patients with pure genotypes to see whether distinct disease associations may emerge.We have been interested to examine these and other common conditions of aging to look for potential associations, and ‘big data’ have been extremely helpful in providing clear answers. These answers have sometimes been surprising. Since AMD and Alzheimer’s disease share many pathological features, it has often been assumed that they are associated in populations. The general conclusion we draw from these studies is that degenerative diseases of aging such as AMD, glaucoma, Alzheimer’s disease and osteoarthritis often share disease mechanisms like inflammation and complement activation, but the underlying ways in which these pathways are triggered may be distinct and dependent on the specific genetic basis of each condition. It is essential that we look to the genetics of each disease in order to understand the biochemical pathways involved and to develop effective treatments. This is particularly true for AMD, where genetics play such a strong role.
How far have we come?
It is vital to realize that that the wide umbrella of the common and debilitating condition of AMD comprises at least two partially distinct entities. Each of these has its own clinical features, biochemical pathways and potential therapies. For chromosome 1 disease, we have now established direct links between the major epidemiological risk factors for AMD – age, genetics and smoking – specific molecular features in human macular tissue. This paves the way for trials of complement inhibitors and other therapies. For these trials to succeed, however, we must select the best candidates prospectively on the basis of patient genotype as well as phenotype. Looking to the future, once we have effective drugs for both chromosome 1 and 10 disease, we can use personalized medicine to provide treatments that target the underlying disease processes and at a much earlier stage. The art of medicine endures. However, the fields of medicine and biological science have become increasingly specialized, with individual groups often pursuing clinical research, genetics or biochemistry in splendid isolation from the other disciplines. To make significant progress towards treating a disease, I feel that we must pursue these approaches in parallel. Perhaps the Greek model of a trireme is useful (Figure 3). As the dominant warships in Ancient Greece, these fast and agile vessels were powered by three rows of oarsmen working in unison to achieve impressive speeds and distances. Could clinical researchers, geneticists and biochemists work in unison like this, with discoveries in epidemiology and genetics used to power and direct biochemical work, creating new therapies to take back to the clinic? In this craft, there may even be a place for the clinician-scientist as κυβερνήτης (helmsman), πρῳρεύς (bow look-out) and occasional rower.
Tiarnán Keenan, is a clinical ophthalmologist and research scientist based at the University of Manchester, UK, with a special interest in age-related macular degeneration and retinal disease.
References
- E Chong, et al., “Age-related macular degeneration phenotypes associated with mutually exclusive homozygous risk variants in CFH and HTRA1 genes”, Retina, 35, 989–998 (2015). PMID: 25627090. TD Keenan, et al., “Assessment of proteins associated with complement activation and inflammation in maculae of human donors homozygous risk at chromosome 1 CFH-to- F13B”, Invest Ophthalmol Vis Sci, 56, 4870–4879 (2015). PMID: 26218915. TD Keenan, et al., “Age-dependent changes in heparan sulfate in human Bruch’s membrane: implications for age-related macular degeneration”, Invest Ophthalmol Vis Sci, 55, 5370–5379 (2014). PMID: 25074778. SJ Clark, et al., “Impaired binding of the agerelated macular degeneration-associated complement factor H 402H allotype to Bruch’s membrane in human retina”, J Biol Chem, 285, 30192–30202 (2010). PMID: 20660596. TD Keenan, et al., “Associations between agerelated macular degeneration, Alzheimer disease, and dementia: record linkage study of hospital admissions”, JAMA Ophthalmol, 132, 63–68 (2014). PMID: 24232933. TD Keenan, et al., “Associations between age-related macular degeneration, osteoarthritis and rheumatoid arthritis: record linkage study”, Retina, Epub ahead of print (2015). PMID: 25996429.
