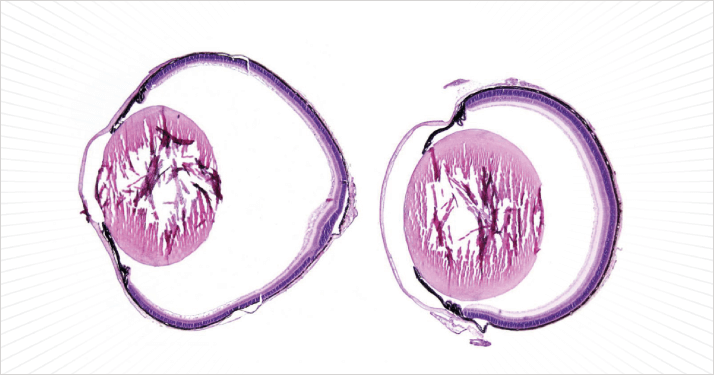
Buphthalmos (abnormal enlargement of the eyes) is often observed in congenital glaucoma, and can result in severe myopia. Although the first recorded mention of the disorder appears to have been in Ancient Greece by Celsius over 2,000 years ago, the normal physiological mechanism that prevents it from developing has remained something of a mystery. But now, a team has uncovered a genetic mechanism which can account for uncontrolled retinal proliferation and eye growth: mutation of the low density lipoprotein receptor-related protein 2 (LRP2) gene. Researchers, including a team from the Max Delbrück Center for Molecular Medicine, Berlin, Germany, found that the developmental morphogen Sonic Hedgehog (SHH) stimulates retinal stem cells to proliferate and differentiate, and the LRP2 receptors found at the outermost margin of the retina act to intercept, degrade and clear it. This keeps the stem cells at the edge of the retina unstimulated, and prevents unrestricted growth of the eye. In mice with mutated LRP2 though, SHH at the periphery is left unchecked, and makes its way to the edge of the retinal margin and stimulates the cells there, which results in significant growth of the eye (1) (see Figure 1).
The findings may also apply to LRP2-deficient patients – large eyes and holoprosencephaly (malformation of the forebrain) are both found in patients with Donnai-Barrow syndrome, a genetic disease resulting from LRP2 mutation (2,3). Both of these phenotypes were also observed in the mouse model, suggesting cross-species conservation of the receptor – and potentially shedding light on an important development mechanism within the human eye.
References
- A Christ, et al., “LRP2 acts as SHH clearance receptor to protect the retinal margin from mitogenic stimuli”, Dev Cell, 35, 36–48 (2015). PMID: 26439398. BR Pober, et al., “A review of Donnai-Barrow and facio-oculo-acoustico-renal (DB/FOAR) syndrome: clinical features and differential diagnosis”, Birth Defects Res A Clin Mol Teratol, 85, 76–81 (2009). PMID: 19089858. JA Rosenfeld, et al., “Clinical characterization of individuals with deletions of genes in holoprosencephaly pathways by aCGH refines the phenotypic spectrum of HPE”, Hum Genet, 127, 421–440 (2010). PMID: 20066439.
