
Basic science; The impact of change in the blood-retinal barrier
Educational content provided by Alimera Sciences.
According to the World Health Organization (WHO), in 2000 there were 170 million people with diabetes mellitus and this number is predicted to rise to an estimated 366 million by 2030. The latest estimates indicate that there were 382 million with diabetes worldwide in 2013, and this number is expected to rise to 592 by 2035.1 Diabetic retinopathy (DR) is a microvascular complication of both type 1 and type 2 diabetes mellitus, and the increasing number of diabetes patients will have an impact on the practice and delivery of ophthalmology services. WHO estimates that DR is responsible for 4.8% of the 37 million cases of blindness throughout the world, and is a leading cause of new-onset blindness in many industrialized countries.2
What are DR and diabetic macular edema (DME)?
The vascular system of the retina is complex, and the retinal neural tissue is protected from potentially harmful molecules in circulation, and excess fluid by the blood-retinal barrier (BRB).3 Hyperglycemia — often seen in diabetes — causes damage to blood vessels and the BRB, and results in increased vascular permeability.4 This causes fluid and molecules to leak into the retina, with extracellular accumulation of fluid and the deposition of macromolecules, leading to the development of retinopathy. Diabetic maculopathy is a condition that can result from retinopathy and consists of damage to the macula, the part of the retina which provides us with our central vision. A common form of damage is from diabetic macular edema (DME) in which fluid builds up on the macula.5,6 DME is characterized by vascular leakage, tissue edema and the deposition of hard exudates in the central retina3 (Figures 1 and 2).
DME is the major cause of vision loss in patients with DR.3 Although DME can occur at any stage of DR, research suggests that the risk of DME rises as the severity of DR increases.7
Figure 1. Early and late frames of fluorescein angiography (left and right, respectively) from a patient with DME in the left eye. Images demonstrate the pooling of extravasated fluorescein due to leakage from dilated capillaries and microaneuryms in the affected retinal area.8
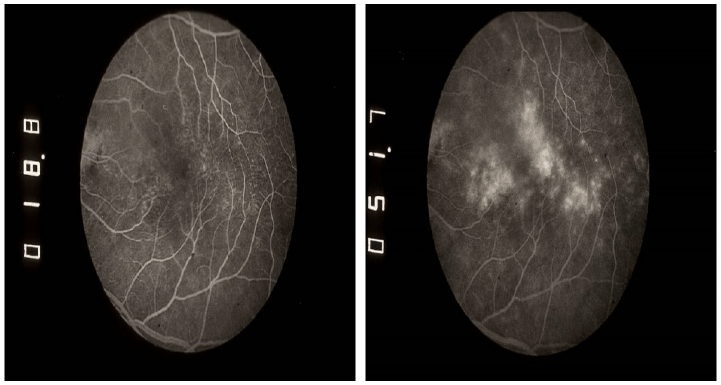
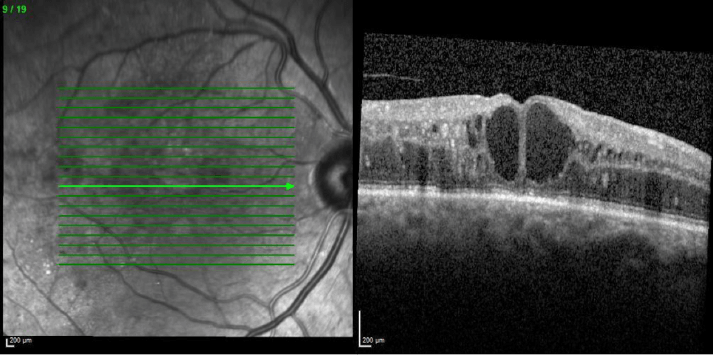
Figure 2. Optical coherence tomography of a patient with DME showing cystoid macular edema recognized by cystic spaces of varying sizes.8
Despite hyperglycemia being the major risk factor for DR, additional risk factors include: genetics, gender, duration of diabetes, hypertension, hyperlipidemia and renal impairment.5,8

Anatomical changes in the BRB
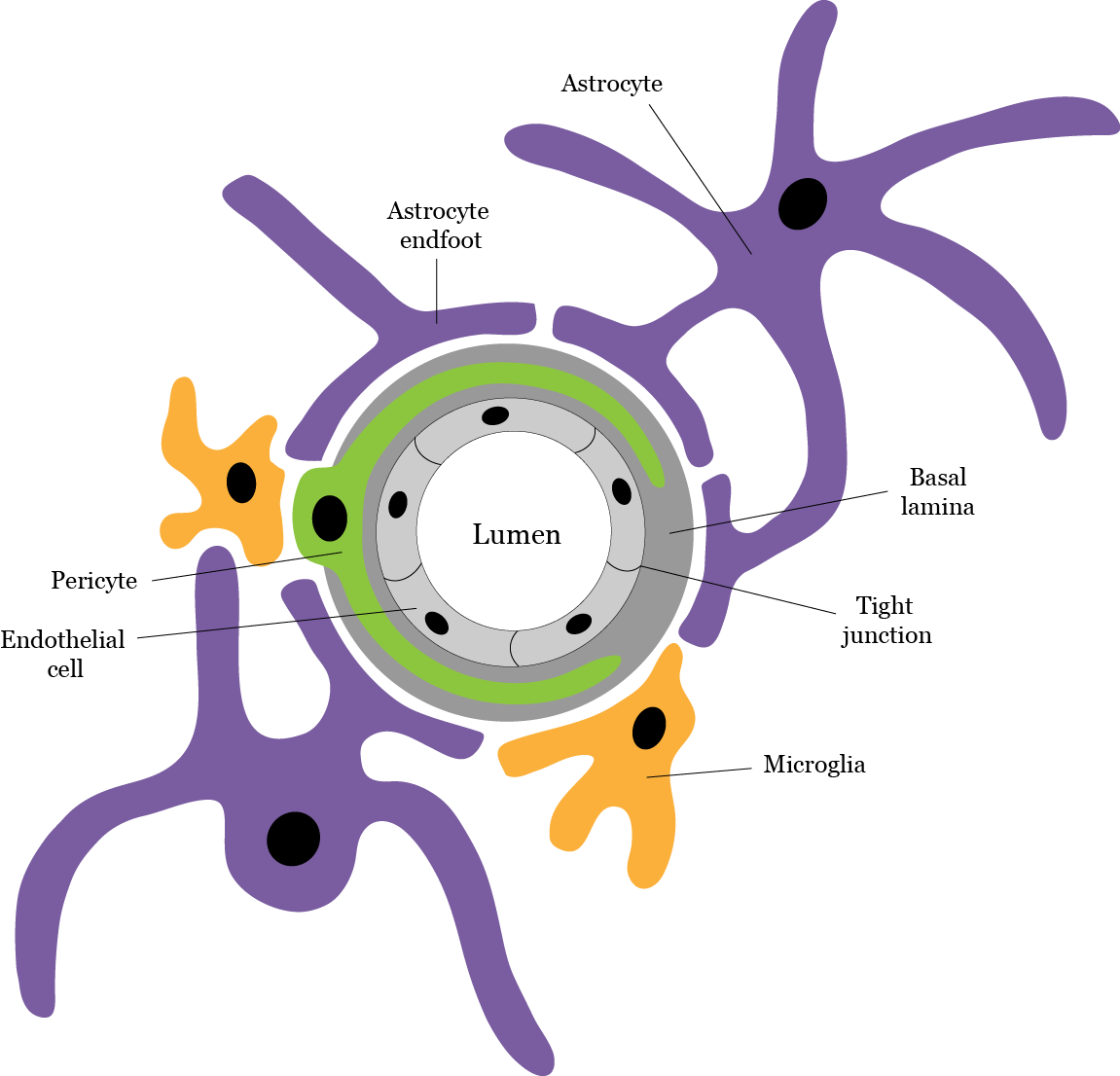
The retinal capillaries that form the BRB consist of a single layer of tightly adherent endothelial cells, a basal lamina and surrounding pericytes, which are enveloped by the processes of astrocytes and microglia. The complex formed by these structures is called the neurovascular unit (Figure 3).3 In healthy individuals, transport of molecules across the BRB is selectively regulated by two pathways: one involving the opening and closing of interendothelial ‘tight’ junctions (paracellular pathway), and another one involving the transport vesicles that travel through the endothelial cells (transcellular pathway).3 Changes in these components can cause the BRB to become leaky and unregulated.
Figure 3. Diagram representing the retinal neurovascular unit, and its components. Adapted from ElAli et al. (2014).
In diabetic individuals, the basal lamina thickens and the endothelial cells have been shown to change shape becoming significantly thinner. It is thought that adhesion of leukocytes to the endothelial cells causes capillary occlusion and BRB breakdown in the early stages of DR.4 After a period of time, the capillaries become acellular due to the loss of pericytes and endothelial cells, and microaneurysms develop. The retinal capillary and arteriolar occlusions lead to retinal ischemia and hypoxia, which may progress to retinal neovascularization, depending on severity.
Although pericytes provide vascular stability, pericyte apoptosis has been observed in early DR leading to BRB breakdown. The mechanism underlying pericyte apoptosis remains unclear; although the accumulation of stable advanced glycation end products (AGEs) — found abundantly in hyperglycemia — has been suggested as a possible cause.3,4
Astrocytes improve barrier properties by inducing the production of tight junction proteins. Glial cells also secrete neuroactive agents and growth factors such as vascular endothelial growth factor (VEGF).3,4 Phenotypic alterations of astrocytes and other glial cells are caused by AGEs formed under hyperglycemic conditions, and result in increased vasopermeability and VEGF induction, which further increase permeability.4
The molecular effect of hyperglycemia on the BRB
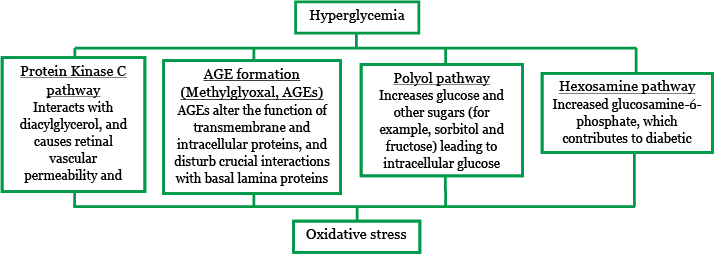
Hyperglycemia causes overproduction of reactive oxygen species in the mitochondria, which leads to oxidative stress and tissue damage through a number of major mechanisms (Figure 4). These mechanisms include:10
- Increased flux of glucose and other sugars through the polyol pathway.
- Increased intracellular formation of AGEs and increased expression of the receptor for AGEs (RAGE).
- Activation of the protein kinase C isoforms.
- Overactivity of the hexosamine pathway.
Figure 4. Hyperglycemia leads to oxidative stress and impacts distinct pathways.8,10,11,12
Oxidative stress leads to pro-inflammatory responses, including the production of inflammatory cytokines such as interleukin (IL)-6, IL-8, IL-1β and tumor necrosis factor-alpha (TNF-α).3 Increased VEGF production and up- and downregulation of other growth factors are also observed.3 In addition, the following have been seen to occur:
- Endothelial dysfunction from AGE and RAGE activation, which increases production of vascular and intracellular adhesion molecules (VCAM-1 and ICAM-1).
- Increased angiopoietin (ANG)-2 and decreased platelet-derived growth factor (PDGF).
- Reduced endothelial cell proliferation.
The molecular changes caused by oxidative stress ultimately result in neurovascular damage and BRB breakdown (Figure 5), leading to macular edema.8 The tight junctions between the endothelial cells in the neurovascular junctions become permeable, and allow small molecules and water into the retina. There is also increased transcellular permeability which allows passage of large molecules and water.
Figure 5. Oxidative stress leads to BRB breakdown and macular edema. Adapted from Winfried Amoaku Pathophysiology of DME presentation.8,12

DME therapeutic approaches

To prevent or limit the progression of DR and DME, patients are advised to work to achieve glycemic control. However, many patients find this difficult to achieve and the eventual development of DR results in visual problems. Laser photocoagulation was the standard for treatment of DME for many years, as it reduces the risk of visual loss and works over a long timescale. However, it is noted that recovery of vision is hard to achieve with laser alone, especially in eyes where the DME involves the centre.5
Intravitreal VEGF inhibitors are also available, and act to reduce VEGF levels in the eye and, thus, reverse the vascular permeability increase. However, as the effects of these anti-VEGF therapies are short lived, the DME may recur. Furthermore, not all eyes with DME are responsive to anti-VEGF therapies, especially if the leakage is chronic.
Corticosteroids are also a treatment option, as they inhibit increased levels of vascular permeability factors described above (including VEGF), and the production and effects of inflammatory mediators, thereby reversing the loss of endothelial tight junction proteins. Intravitreal steroid treatment has been shown to be effective in the treatment of DME, including cases where there is suboptimal response to anti-VEGF therapies.5
The available pharmacological treatments for DME — that is, intravitreal steroids and anti-VEGFs — can be used with or without laser treatment in order to achieve optimal outcomes.5
Winfried Amoaku has provided consultancy services to Alcon, Alimera, Allergan, Bayer, Novartis and Thrombogenics. He has received travel grants from Alimera, Allergan, Bayer and Novartis, and honoraria for lectures from Allergan and Novartis. He has participated in clinical trials for which his institution has received funding from Allergan, Novartis and Pfizer. His institution has further received research grants from Allergan and Novartis for non-clinical studies, and CentreVue (Italy) for clinical studies.
REFERENCES
1. L Guariguata et al., “IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2013 and projections for 2035”, Diabetes Res Clin Pract, 103, 137–149 (2014).
2. World Health Organization, “Prevention of blindness from diabetes mellitus”, Report of a WHO consultation in Geneva, Switzerland, 9–11 November 2005.
3. I Klaassen et al., “Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions”, Prog Retin Eye Res, 34, 19–48 (2013). PMID: 23416119.
4. N Bhagat et al., “Diabetic macular edema: pathogenesis and treatment”, Surv Ophthalmol, 18, 116–120 (2003). PMID: 15513471.
5. The Royal College of Ophthalmologists, “Diabetic retinopathy guidelines”, (2012, updated 2013). Available at: www.rcophth.ac.uk/standards-publications-research/clinical-guidelines/. Accessed July 2015.
6. Diabetes UK. Diabetic Maculopathy. Accessed July 2015. www.diabetes.co.uk/diabetes-complications/diabetic-maculopathy.html
7. M Henricsson et al., “Progression to proliferative retinopathy and macular oedema requiring treatment. Assessment of the alternative classification of the Wisconsin study”, Acta Ophthalmol Scand, 77, 218–233 (1999). PMID: 10321543. (onlinelibrary.wiley.com/doi/10.1034/j.1600-0420.1999.770221.x/epdf).
8. W Amoaku, “Pathophysiology of diabetic macular oedema”. Presentation at the RCOphth Annual Congress Liverpool, UK; 19–21 May 2015.
9. A ElAli et al., “The role of pericytes in neurovascular unit remodeling in brain disorders”, Int J Mol Sci, 15(4), 6453–6274 (2014). PMID: 24743889.
10. F Giacco, M Brownlee, “Oxidative stress and diabetic complications”, Circ Res, 107, 1058–1070 (2010). PMID: 21030723.
11. X Zhang et al., “Diabetic macular edema: new concepts in patho-physiology and treatment”, Cell Biosci, 4, 27 (2014). PMID: 24955234.
12. A Das et al., “Diabetic Macular Edema: Pathophysiology and Novel Therapeutic Targets”, Ophthalmology, 122, 7, 1375–1394 (2015). PMID: 25935789.
UK-ILV-MMM-0280
Date of preparation: July 2015
Founded in 2003, Alimera Sciences researches and develops innovative vision-improving treatments for chronic retinal diseases, such as diabetic macular edema (DME), dry age-related macula degeneration (AMD), and retinal vein occlusion. In 2015, Alimera Sciences partnered with The Ophthalmologist to facilitate the publication of independently created educational content surrounding DME, a serious retinal complication associated with diabetes, which is increasing in incidence with the increasing prevalence of diabetes worldwide. Published content will include articles ranging from basic science and disease processes to overviews of clinical data, different surgical procedures, comparisons of treatment options, and practical advice for managing diabetic patients. With a commitment to honesty, integrity, responsibility, candor, and trust, Alimera Sciences intend to provide educationally focused content to healthcare professionals across a wide range of topics in DME in order to both increase disease awareness and understanding, and to help improve patient outcomes. UK-ILV-MMM-0355 Date of preparation: August 2015 enquiries@alimerasciences.com
